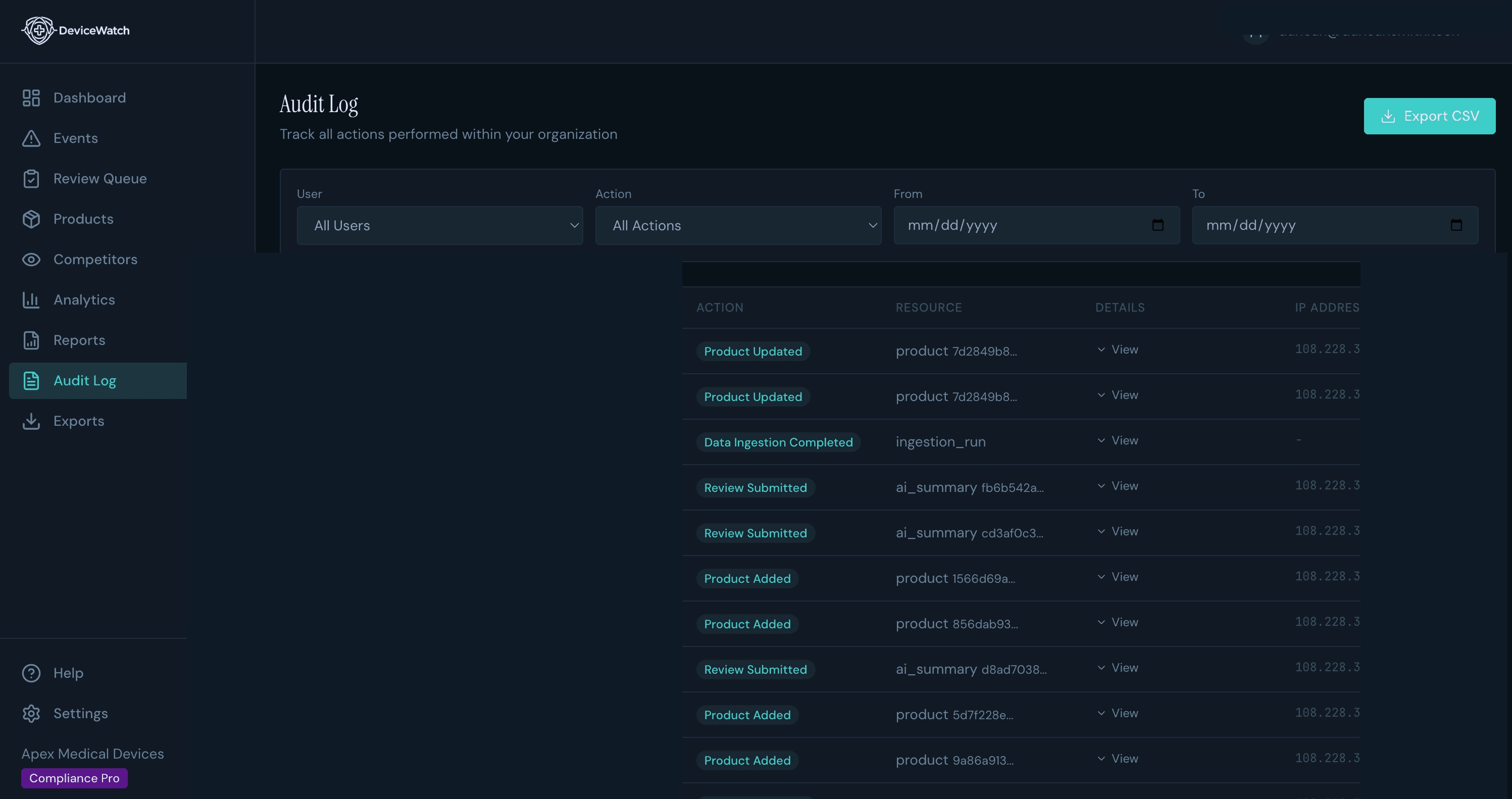
Audit-grade surveillance your quality team can defend.
DeviceWatch monitors FDA adverse events for your product codes, summarizes clinical narratives with AI, and logs every review decision to a tamper-evident, hash-chained audit trail with 21 CFR Part 11 e-signatures. When a notified body asks for proof, you already have it.
Built for regulated environments
Public FDA data only. Zero PHI.
Built on public regulatory data via the openFDA API — zero patient-identifiable information ever enters the system.
21 CFR Part 11 e-signatures
Part 11-aligned audit trails with e-signature capture on every review decision your team signs off.
Hash-chained, tamper-evident audit trail
Every review action, login, and export logged with timestamps, user attribution, and integrity verification.
Watch the 2-Minute Demo
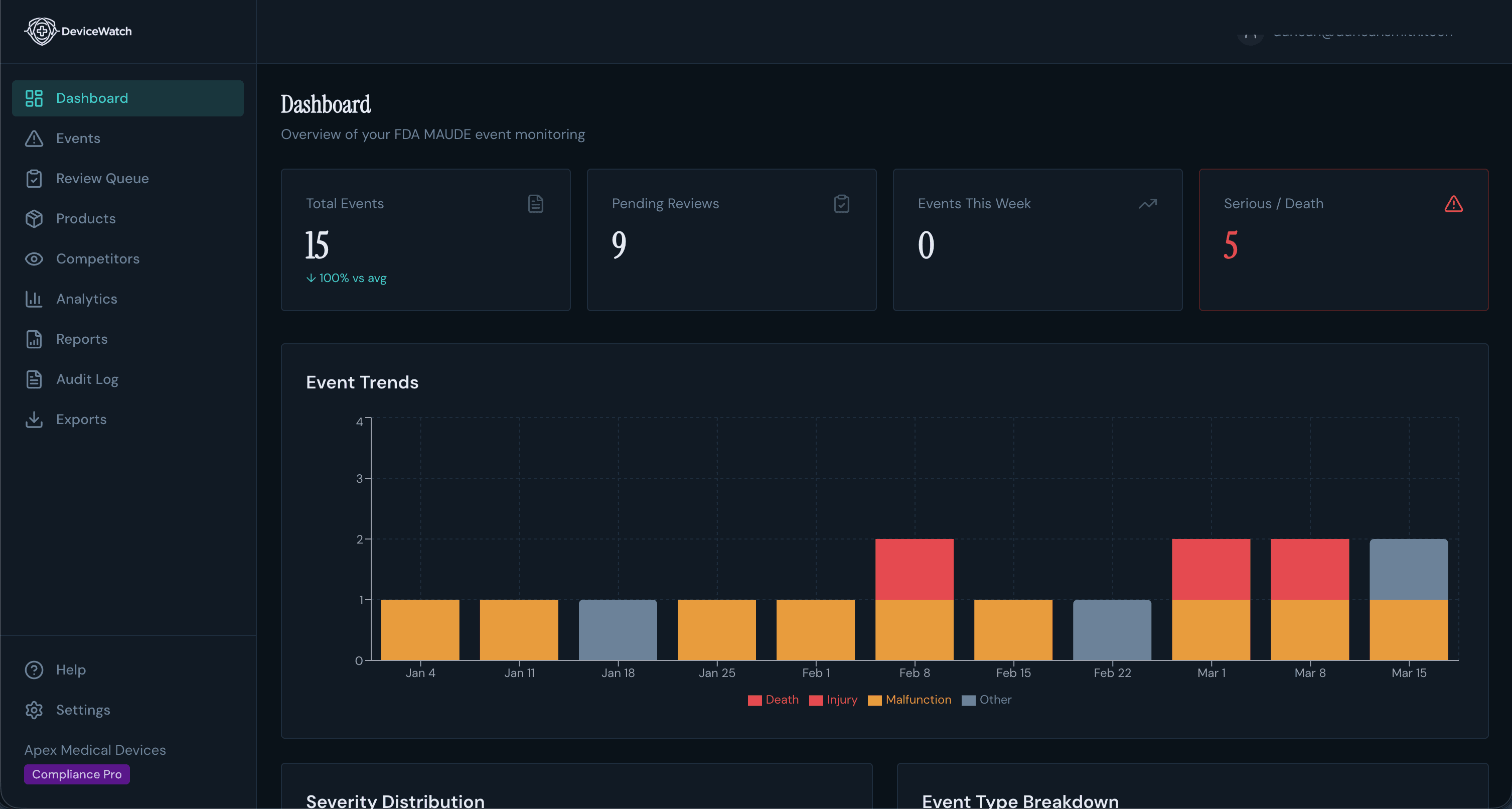
See DeviceWatch Read 2,000 Reports a Day
From raw FDA AEMS (formerly MAUDE) narratives to reviewed, audit-ready intelligence — the whole pipeline in under two minutes.
- 0:00The 2,000-reports-a-day problem
- 0:40Automated AEMS ingestion
- 0:53AI summaries + human review
- 1:20Signals, recalls & 510(k)
- 1:40Part 11 audit & one-click reports
The Problem
The Data Is Growing Faster Than Any Team Can Read It
The cost of missing a signal isn't a fine. It's a recall.
Everything You Need
Your Team Makes Decisions. DeviceWatch Does Everything Else.
Not another eQMS. A purpose-built surveillance engine that feeds your existing quality system better data, faster, from FDA adverse events to audit-ready compliance documents.
Never Search AEMS Again
Add your product codes once. DeviceWatch pulls every new adverse event from FDA AEMS (formerly MAUDE) automatically, so your regulatory team stops wasting hours on data entry and starts focusing on patient safety.
See What Competitors Can't Hide
Track adverse events on competing devices alongside your own. When a regulator asks "how does your safety profile compare?", you'll have the answer ready.
More monitor capabilities (2)
Critical Signals, Not Noise
Severity-filtered alerts hit your Slack, Teams, or webhooks instantly. Death and serious injury events surface immediately while minor malfunctions wait for your weekly digest.
Your Weekly Intelligence Briefing
Every Monday: a formatted safety signal digest grouped by severity, ready to forward to leadership. What used to take your team a full day now arrives in your inbox automatically.
AI That Reads 10,000 Narratives So You Don't Have To
Each adverse event report contains dense clinical narrative buried in FDA formatting. Claude AI extracts the failure mode, severity, root cause, and patient outcome in seconds, not hours.
AI Finds It. You Verify It.
No black-box automation. Every AI-generated summary lands in a human review queue where your team acknowledges, flags, or rejects each finding. Full audit trail included.
More analyze capabilities (2)
From Raw Data to Boardroom-Ready Charts
Interactive dashboards show event frequency, severity trends, and emerging failure patterns over time. Share a link with leadership instead of building another slide deck.
Signal Detection Engine
Our automated anomaly detection identifies volume spikes, shifting severity patterns, and emerging failure clusters before they become regulatory findings, so you escalate proactively, not defensively.
PSURs That Used to Take Weeks Now Take Days
Auto-generated periodic safety update reports with trend analysis, signal summaries, and data pre-formatted for regulatory submission. Your team reviews and signs off. The grunt work is done.
CAPA Documentation Without the Data Hunt
Corrective action templates pre-populated from your adverse event data. Start documenting the moment a signal is confirmed, not after days of gathering evidence from scattered spreadsheets.
More act capabilities (2)
Audit-Ready in One Click
When your notified body asks for post-market surveillance evidence, you export a compliance-ready PDF from your own custom report templates — backed by a hash-chained, integrity-verifiable audit log. No scramble to compile one. Formatted for FDA submissions, EU MDR audits, or internal review.
Your Data, Your Workflows
Full REST API to feed surveillance data into your existing QMS, build custom dashboards, or trigger workflows in tools your team already uses. No vendor lock-in.
See It In Action
Built for Regulatory Professionals
Every screen designed around the workflows your team actually uses.
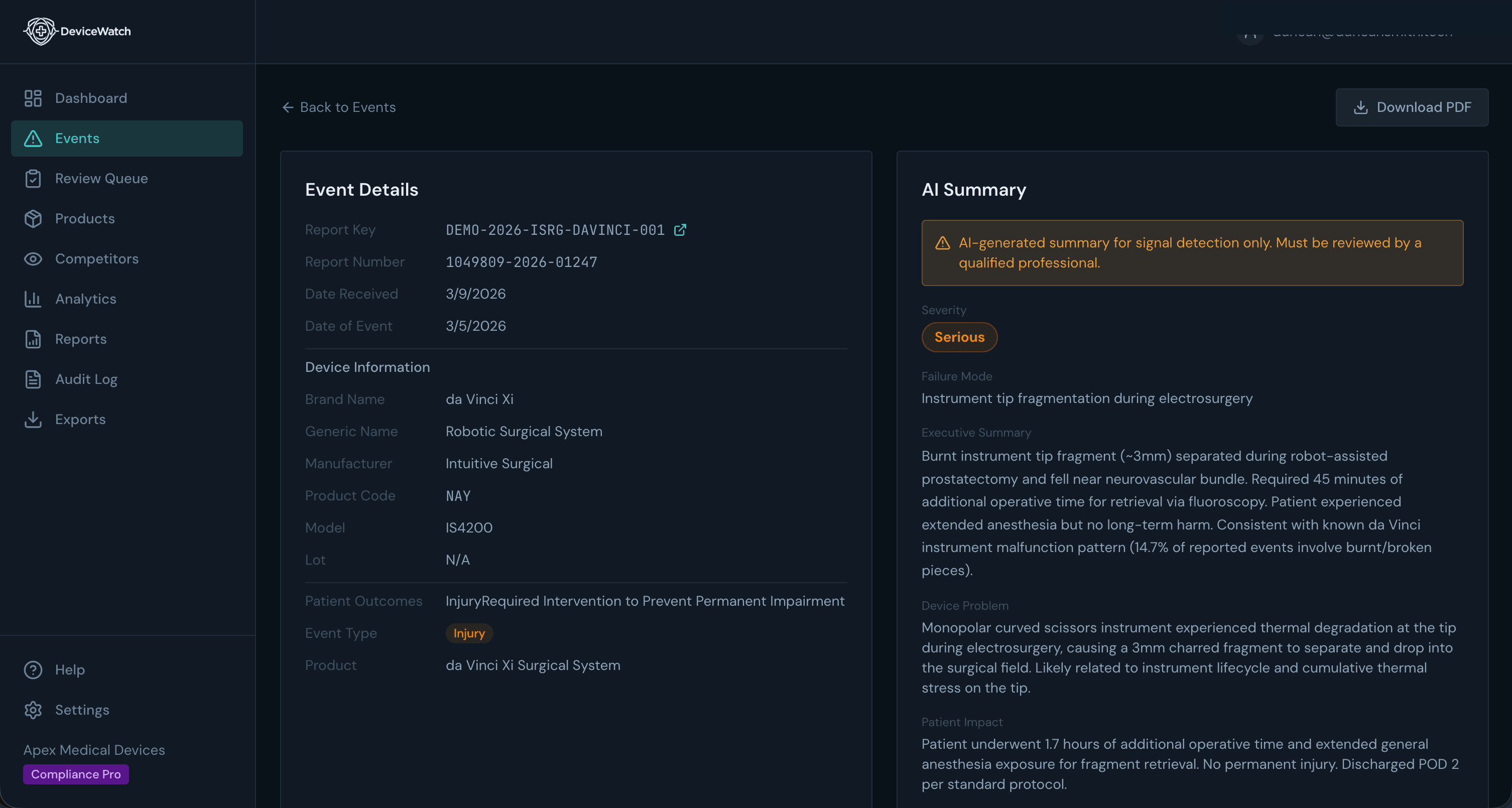
AI-Powered Analysis
Every Event, Summarized in Seconds
Dense FDA narratives distilled into actionable summaries. Device information, failure modes, severity, patient outcomes — extracted automatically, reviewed by your team.
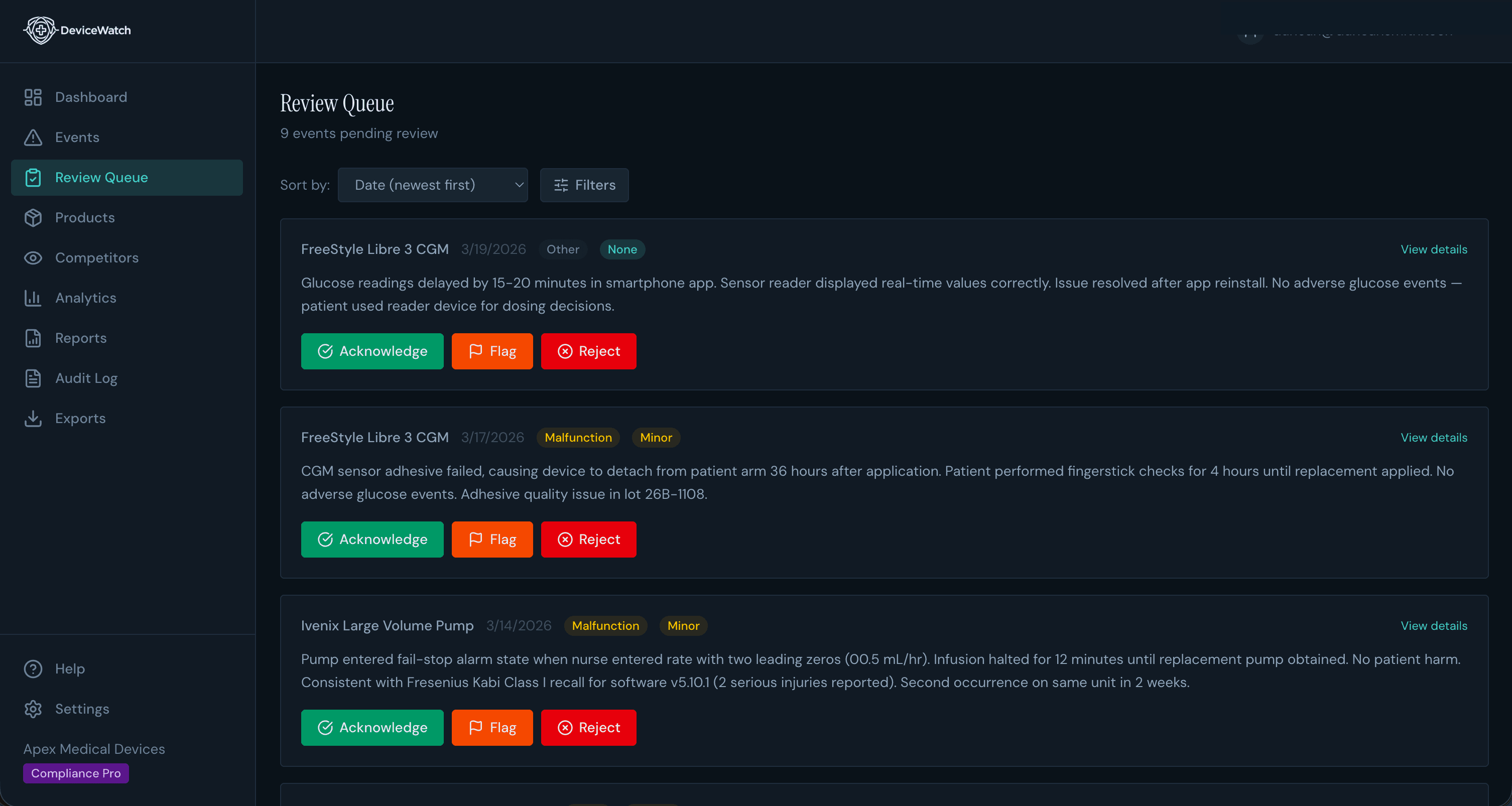
Human-in-the-Loop
AI Finds It. You Verify It.
No black-box automation. Every AI summary lands in a review queue where qualified professionals acknowledge, flag, or reject findings — captured with Part 11 e-signatures and a hash-chained audit trail at every step.
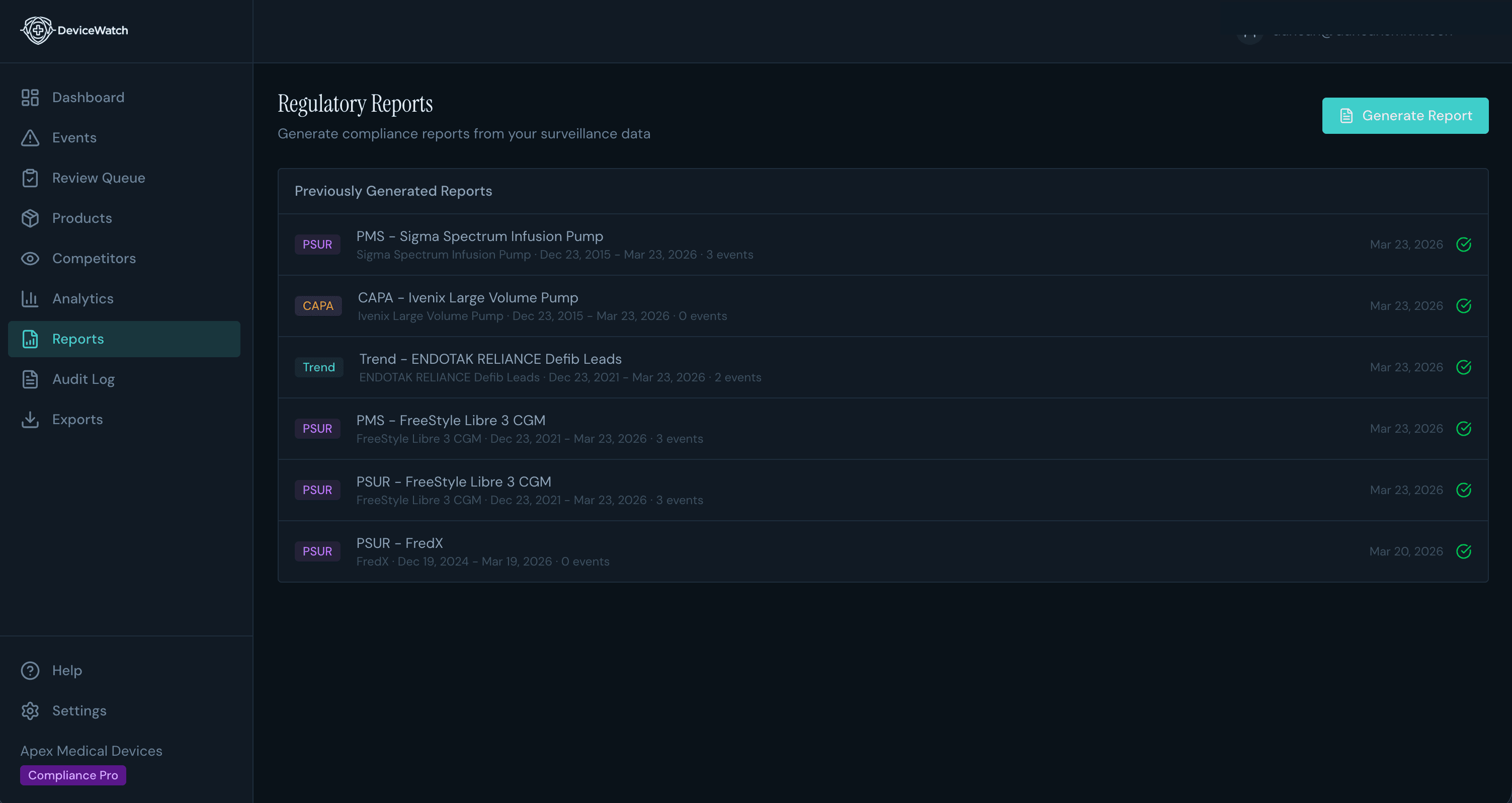
Compliance-ready regulatory reports
21 CFR Part 11 audit trails
How It Works
From Raw Data to Actionable Intelligence
DeviceWatch ingests global adverse event data, applies AI analysis, and delivers compliance-ready insights to your team.
Data Sources
Harness Data
openFDA, AEMS Dashboard
Track Recalls
FDA Recalls, MHRA UK
International Events
Global vigilance monitoring
Clinical Literature
PubMed scanning
AI Intelligence Engine
Signal Detection
Failure Clustering
AI Summarization
Competitor Analysis
Trend Forecasting
Literature Matching
Output & Action
Review Queue
Prioritize critical issues
PSUR & PDF Reports
Automate regulatory compliance
Instant Alerts
Slack, email notifications
API Integration
Seamless system connectivity
Competitive Intelligence
Analyze your performance vs competitors
Compare failure modes & trends across devices
Optimize strategy with adverse event market insights
ROI Calculator
What Manual Surveillance Actually Costs You
Manual cost/yr
$187,200
DeviceWatch/yr
$5,988
Annual savings
$181,212
ROI
3,026%
Default hours based on Greenlight Guru 2025 State of Medical Device Industry Report (n=536). DeviceWatch Growth plan at $499/mo.
Pricing
Less Than the Cost of One Manual PSUR Cycle
14-day free trial on every plan. No credit card. Cancel anytime.
Growth
Competitive intelligence, full analytics, and reports that used to take weeks, for less than one consultant day.
- 15 product codes
- Competitor device tracking
- Full analytics suite
- Team collaboration
- PSUR-ready trend reports
- FDA recalls & 510(k) clearances
- Slack & Teams alerts
- PDF exports
- Custom alert rules
- Priority support
Compliance Pro
Enterprise-grade compliance infrastructure with Part 11-ready audit trails, unlimited scale, and dedicated support.
- Unlimited product codes
- Hash-chained Part 11 audit trails
- Safety signals & CAPA tracking
- E-signature capture (Part 11)
- API access
- Unlimited team members
- Automated PSUR reports
- Custom report templates & branding
- Multi-source surveillance
- Custom webhooks
- Dedicated account manager
- SLA guarantee
- Custom integrations
Enterprise
Dedicated infrastructure, global regulatory coverage, and full validation packages for large-scale medical device organizations.
- Everything in Compliance Pro
- Part 11-ready + EU MDR/IVDR audit trails
- Automated PSUR, CER & PMCF reports
- SSO/SAML + SCIM provisioning
- Dedicated instance or on-prem deployment
- 24/7 support + named Customer Success Manager
- IQ/OQ/PQ validation package included
- Custom data retention & residency
- Unlimited API access + bulk export
- Annual contract with BAA/MSA
FAQ
Questions Before You Start
What exactly does DeviceWatch replace in our workflow?
The hours your team spends manually searching FDA AEMS, reading clinical narratives, copying data into spreadsheets, and compiling it into reports. DeviceWatch automates the entire pipeline: ingestion, AI analysis, human review, and report generation. Your team reviews and makes decisions. The data gathering is done.
How does the AI analysis work, and can we trust it?
We use Claude by Anthropic to read dense clinical narratives and extract failure modes, severity classifications, patient outcomes, and root cause indicators. But here's what matters: every AI summary lands in a human review queue. Your qualified professionals acknowledge, flag, or reject each finding before it enters your compliance record. The AI accelerates the work; your team owns the decisions.
Is this compliant with 21 CFR Part 11?
DeviceWatch is built for Part 11 environments: immutable audit trails, role-based access controls, session timeouts, data integrity verification, and exportable audit logs, built in from day one. Every review action, login, and export is logged with timestamps and user attribution. Part 11 compliance ultimately rests on your validated procedures; DeviceWatch gives you the technical controls and the documentation trail to support them.
Why would I track competitor devices?
Because regulators compare your safety profile to the market. When your notified body asks "how does your adverse event rate compare to similar devices?", you'll have the data ready. Spot industry-wide failure patterns early, benchmark your device's performance, and know what questions are coming before the audit.
How much time will this actually save our team?
The manual PSUR data preparation that takes 2-3 weeks? DeviceWatch cuts it to 2-3 days. The recurring FDA adverse-event search that eats an afternoon? Gone — new events stream in automatically the moment they publish. But the real ROI is strategic: your regulatory affairs team stops being data clerks and starts being the safety experts you hired them to be.
We already have a QMS. How does this fit?
DeviceWatch doesn't replace your QMS. It feeds it better data, faster. Your QMS manages the full quality lifecycle (CAPAs, NCRs, design controls). DeviceWatch is laser-focused on post-market surveillance: automated FDA adverse-event monitoring, AI-powered analysis, signal detection, and regulatory report generation. The API lets you pipe findings directly into your existing quality workflows.
What data sources does DeviceWatch monitor?
Today: FDA adverse events (via AEMS, including the full MAUDE archive), FDA device Recalls, and 510(k) clearances — all via the openFDA API, ingested the moment new records publish and matched to your monitored product codes. On the roadmap: MHRA (UK), BfArM (Germany), PubMed clinical literature, and EUDAMED when available. All public regulatory data, zero PHI, zero patient-identifiable information.
What about FDA's new AEMS database? Is MAUDE being replaced?
The FDA launched its Adverse Event Monitoring System (AEMS) in March 2026 and completed the migration of all MAUDE data by May 2026. AEMS now contains device adverse events and the full MAUDE archive. DeviceWatch monitors AEMS through the openFDA API and ingests new events the moment they publish. You change nothing — we handle the data source transition server-side.
Can I try it before committing?
Every plan includes a 14-day free trial. No credit card required. Add your product codes, see your first AI-analyzed events within minutes, and generate a sample report. If it doesn't save your team time, you've lost nothing.
TrendWatch
The safety signals you wouldn’t see anywhere else.
A free weekly brief for RA/QA teams: Class I alerts, the adverse-event signals that precede recalls, and one teardown with a CAPA lesson. See what’s inside →
Your Regulatory Team Was Hired to Protect Patients, Not Search Databases.
Give them the tools to focus on what matters. Start your free trial and see your first AI-analyzed adverse events within minutes.